The defensible asset isn't the drug, it's the dossier.
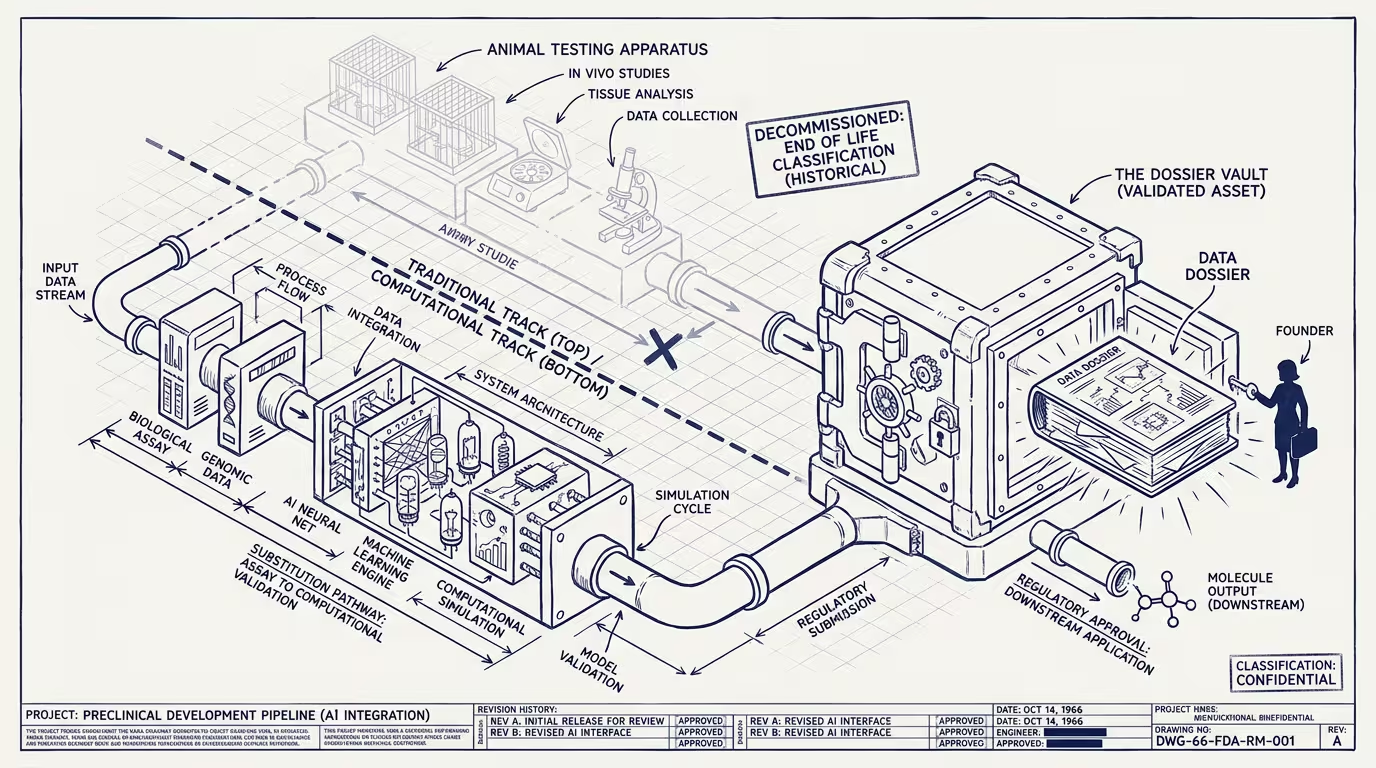
The FDA published a strategic roadmap in early 2025 to reduce animal testing in preclinical drug development. The framing endorsed AI computational models as alternatives where the validation evidence supports the substitution. The roadmap is, in operating terms, the regulatory door opening for a category that has been under-built for two decades because the regulatory frame did not exist.
The asset isn't the drug. _The asset is the dossier._
Hold that frame in view. Every implication that follows traces back to it.
What the dossier is, mechanically: an FDA-validated computational pipeline that produces preclinical safety evidence acceptable for IND-filing. The pipeline ingests molecular structure, predicts ADMET properties (absorption, distribution, metabolism, excretion, toxicity), models known toxicology pathways, and outputs a validation package the agency accepts. The molecule the pipeline tested is replaceable. The next molecule runs through the same pipeline. The pipeline is, in operating terms, the durable competitive moat. The biotech that owns the validated pipeline can run candidates through it at marginal cost. The biotech that does not has to either license the pipeline (paying rents) or run animal studies (paying the legacy cost structure).
What changes is what counts as preclinical safety evidence. Through the post-1962 regulatory era, preclinical safety evidence was animal-assay results: rodent, dog, primate studies showing that the candidate molecule did not produce unexpected toxicity. The mechanism was biological. The cost was high (a typical preclinical animal-study program runs $2-10M per candidate). The timeline was long (12-24 months for the assay-to-IND-filing window). The data quality was variable (animal models do not perfectly translate to human biology).
Computational alternatives existed in the literature for a decade and lacked regulatory standing. A founder building a computational-toxicology platform in 2018 was building against an FDA framework that did not yet accept the model output as sufficient evidence. The platform existed. The data was good. The regulator did not write it into the rulebook. The category was under-built because the demand-side validation was not there.
The 2025 roadmap changes the demand-side validation. Computational models, properly validated, can substitute for some animal-assay categories. The substitution does not happen automatically — the model has to clear FDA review on its own merits, the validation evidence has to meet the agency's standards, and the substitution is initially scoped to specific safety endpoints rather than to the full preclinical-evidence package. But the door is open. Operators in computational-toxicology, in-silico ADMET modeling, and broader computational-biology platforms can now build product against an FDA frame that recognizes the output.
Trace it back to the dossier-as-asset frame and the validated-pipeline-builders are the new platform layer of biotech. Companies that get FDA validation on their computational platforms become, in operating terms, what AWS became to cloud computing. The platform is the durable infrastructure. The drug-developer is the customer. The platform's revenue is a function of how many customers run candidates through it; the customer's cost structure improves; the platform's moat compounds with each validation event.
Trace it back again and the timing of validation matters more than the technical quality of the platform. A platform with marginally-best computational accuracy that gets FDA-validated in 2026 captures the category. A platform with technically-best computational accuracy that gets FDA-validated in 2028 enters a market where the 2026-validator owns the dossier-as-asset position. Operators racing to validation are racing for category position, not just for product-quality.
Trace it back once more and the pharma-incumbent class is structurally exposed. Pharma's preclinical infrastructure is calibrated to animal-assay economics. The transition to computational substitutes is operationally hard for the incumbent — the labs, the staff, the regulatory-affairs apparatus all have to retool. The startup-class that builds platform-native runs faster on the new evidence-frame. By 2027-2028 the structural shift is visible in pharma-balance-sheet allocations toward computational-platform partnerships and platform-acquisition activity.
The same shape recurs across other regulated categories. The FDA's evidence-frame shift is going to repeat. Medical-device safety evidence is already in motion. Diagnostic-test validation is in motion. Clinical-trial-design optimization is in early discussion. In each category, the regulatory frame's accommodation of AI-validated evidence creates a platform-layer category that did not previously exist.
What survives all of this is the structural read that the FDA's roadmap is one of the most consequential biotech-category-creation events of the decade, the trade-press will write it up as a regulatory-modernization story rather than as a category-creation story, and the operators who recognize the dossier-as-asset framing in 2025 are the operators who own the computational-platform category in 2030.
The defensible asset isn't the drug. It's the dossier. The platform that owns the dossier owns the next decade of biotech infrastructure. The FDA just opened the door.
—TJ